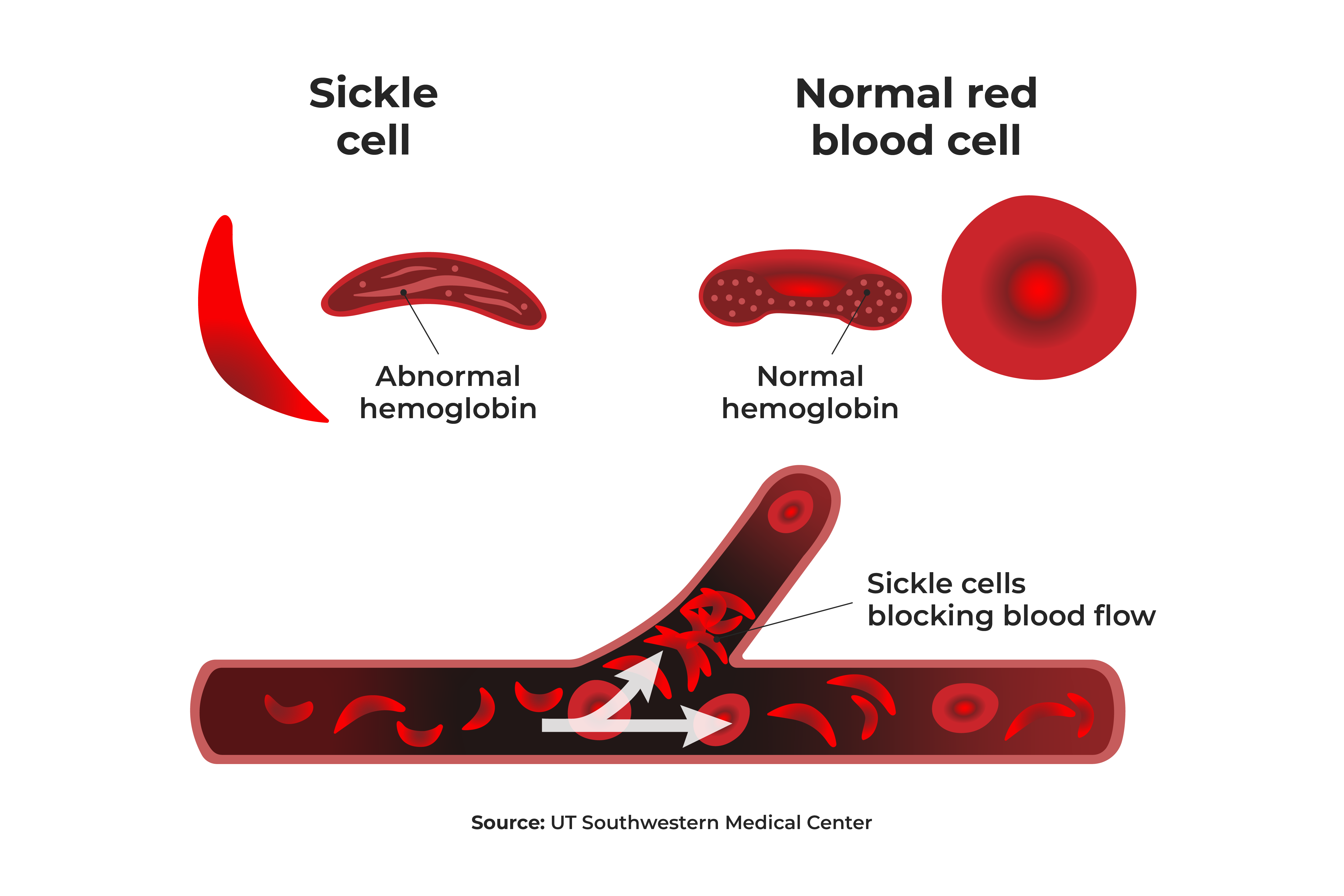
Sickle cell disease (SCD) is the most common inherited blood disorder in the United States, impacting approximately 100,000 Americans. The disease is caused by a gene mutation that causes individuals to produce abnormal hemoglobin, which makes it difficult for red blood cells to carry oxygen from the lungs to other parts of the body. SCD is a lifetime disease, with signs and symptoms usually appearing in infants in their first year.
Normally, red blood cells are flexible and round, allowing them to move easily through the blood vessels. But in SCD, the red blood cells are shaped like crescent moons or “sickles.” The irregularly shaped blood cells become rigid and sticky and can get stuck in the small blood vessels, causing slow or blocked blood flow and reduced oxygen levels, all of which can to organ and tissue damage and episodes of pain crisis.